Welcome to the Sickle Cell Disease Program
In 1972 the Virginia Sickle Cell Disease Center was initiated at MCV with Florence Neal Cooper Smith and Dr. Robert B. Scott at the helm. Today under the leadership of Dr. Wally Smith the program has expanded to include treatment, research and support of patients with Sickle Cell Disease throughout the state of Virginia.
People diagnosed with Sickle Cell Disease live with uncertainty, as crippling symptoms can erupt without warning. Our team offers research-based and evidence-based treatments to help improve your quality of life – and potentially cure your disease. We’re available 24/7 to help when you need us most.
We offer everything you need in one stop: medical inpatient and outpatient treatment, support services to aid in managing day-to-day needs and integrated care with other specialists should complications develop. Beyond that, our doctors develop and test new therapies. Our whole-patient approach means we also help you with the social and mental health impacts of living with a life-long disease – one that isn’t visible to others.
Our clinic
Learn more about the VCU Adult Sickle Cell Adult Medical Home


Welcome
Program Director: Wally Smith, MD
Sickle Cell
Learn more about the disease
Sickle cell disease is a group of inherited red blood cell disorders. People who have sickle cell disease have an abnormal protein in their red blood cells. In the United States, most people who have sickle cell disease are of African ancestry, but the condition is also common in people with a Hispanic background. Because the disease runs in families, couples planning to have children can have genetic testing.
Early signs and symptoms of sickle cell disease include swelling of the hands and feet; symptoms of anemia, including fatigue, or extreme tiredness; and jaundice. Over time, sickle cell disease can lead to complications such as infections, delayed growth, and episodes of pain, called pain crises. Most children who have sickle cell disease are pain-free between crises, but adolescents and adults may also suffer with chronic, ongoing pain. Over a lifetime, sickle cell disease can harm a patient’s spleen, brain, eyes, lungs, liver, heart, kidneys, penis, joints, bones, or skin.
A blood and bone marrow transplant is currently the only cure for sickle cell disease, and only a small number of people who have sickle disease are able to have the transplant. There are effective treatments that can reduce symptoms and prolong life. Early diagnosis and regular medical care to prevent complications also contribute to improved well-being. Sickle cell disease is a lifelong illness. The severity of the disease varies widely from person to person.
People who have sickle cell disease have abnormal hemoglobin, called hemoglobin S or sickle hemoglobin, in their red blood cells. Hemoglobin is a protein in red blood cells that carries oxygen throughout the body. People who have sickle cell disease inherit two abnormal hemoglobin genes, one from each parent.
The types of sickle cell disease include the following:
- Hemoglobin Sβ0 thalassemia
- Hemoglobin Sβ+ thalassemia
- Hemoglobin SC
- Hemoglobin SD
- Hemoglobin SE
- Hemoglobin SS
In all types of sickle cell disease, at least one of the two abnormal genes causes a person’s body to make hemoglobin S. When a person has two hemoglobin S genes (hemoglobin SS), the disease is called sickle cell anemia. This is the most common and often most severe type of sickle cell disease. Hemoglobin SC disease and hemoglobin Sβ thalassemia are two other common types of sickle cell disease. Hemoglobin SD and hemoglobin SE are much less common.
How is the hemoglobin S inherited?
When the hemoglobin S gene is inherited from only one parent, and a normal hemoglobin gene—hemoglobin A—is inherited from the other, that person will have sickle cell trait. People who have sickle cell trait are generally healthy.
Only rarely do people who have sickle cell trait have complications similar to those seen in people who have sickle cell disease. But people who have sickle cell trait are carriers of a defective hemoglobin S gene, so they can pass it on when they have a child.
If the child’s other parent also has sickle cell trait or another abnormal hemoglobin gene, such as beta-thalassemia, hemoglobin C, hemoglobin D, or hemoglobin E, that child has a chance of having sickle cell disease.
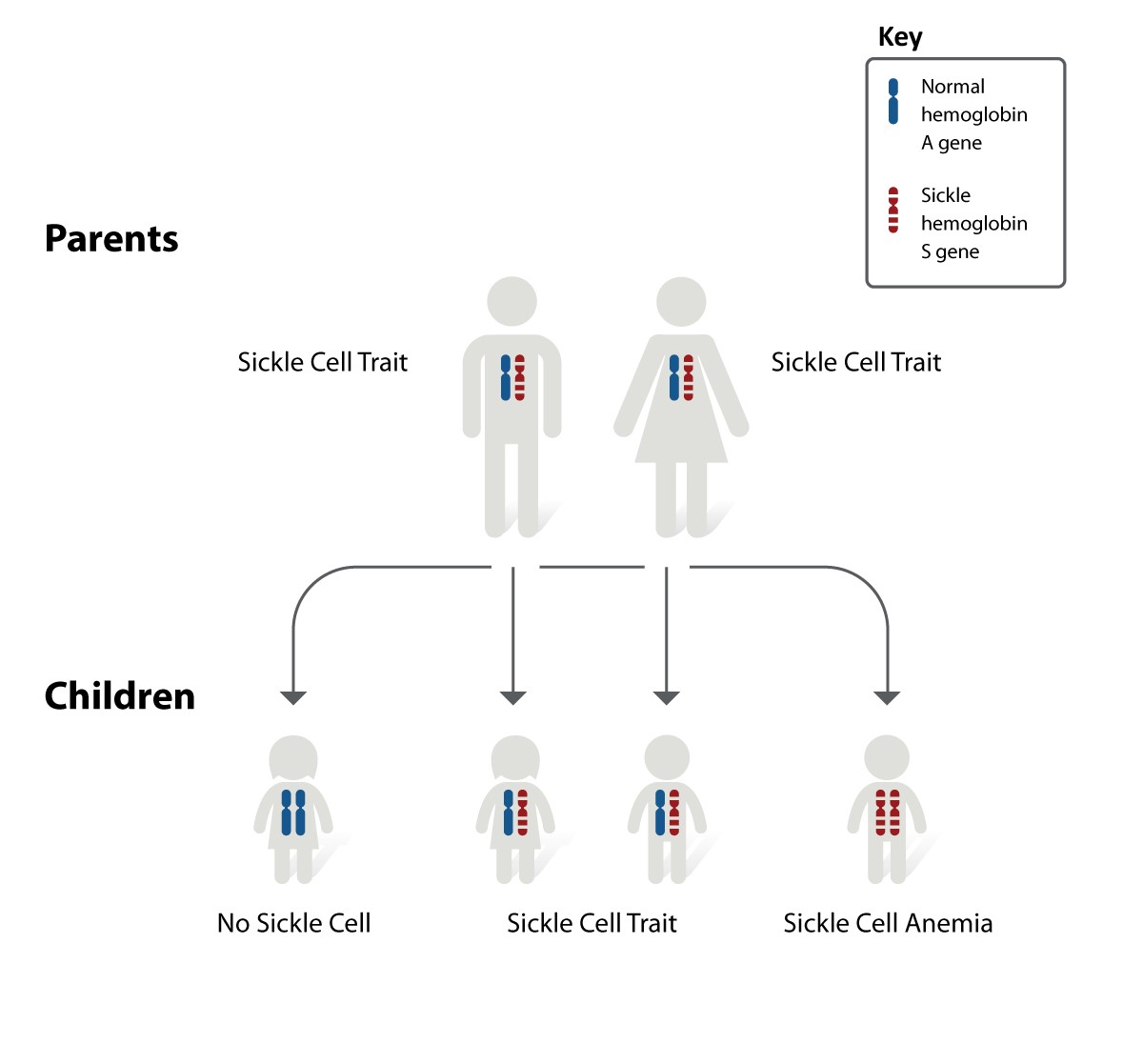
Inheritance pattern for sickle cell disease. The image shows how hemoglobin S genes are inherited. A person inherits two hemoglobin genes—one from each parent. A normal hemoglobin A gene will make normal hemoglobin. A hemoglobin S gene will make abnormal hemoglobin. (https://www.nhlbi.nih.gov/health-topics/sickle-cell-disease)
In the image above, each parent has one normal hemoglobin A gene and one hemoglobin S gene, which means each of their children has:
- A 25 percent chance of inheriting two normal hemoglobin A genes. In this case, the child does not have sickle cell trait or disease.
- A 50 percent chance of inheriting one normal hemoglobin A gene and one hemoglobin S gene. This child has sickle cell trait.
- A 25 percent chance of inheriting two hemoglobin S genes. This child has sickle cell disease.
It is important to keep in mind that each time this couple has a child, the chances of that child having sickle cell disease remain the same. In other words, if the firstborn child has sickle cell disease, there is still a 25 percent chance that the second child will also have the disease. Both boys and girls can inherit sickle cell trait, sickle cell disease, or normal hemoglobin.
If a person wants to know whether he or she carries a sickle hemoglobin gene, a doctor can order a blood test to find out.
Abnormal hemoglobin, called hemoglobin S, causes sickle cell disease. Sickle cell disease is an inherited disease caused by defects, called mutations, in the beta globin gene that helps make hemoglobin. Normally, hemoglobin in red blood cells takes up oxygen in the lungs and carries it through the arteries to all the cells in the tissues of the body. Red blood cells that contain normal hemoglobin are disc-shaped and flexible so that they can move easily through large and small blood vessels to deliver oxygen.
Sickle hemoglobin is not like normal hemoglobin. The mutations in the gene cause a problem when oxygen levels in the blood are lower, which occurs once the hemoglobin has delivered oxygen to the cells in the body’s tissues. With less oxygen, the abnormal hemoglobin S gene can cause rigid, nonliquid protein strands to form within the red blood cell. These rigid strands can change the shape of the cell, causing the sickled red blood cell that gives the disease its name.
Sickle-shaped cells are not flexible and can stick to vessel walls, causing a blockage that slows or stops the flow of blood. When this happens, oxygen is unable to reach nearby tissues. The lack of oxygen in tissue can cause attacks of sudden severe pain, called pain crises. These pain attacks can occur without warning, and a person who has them often needs to go to the hospital for effective treatment.
Because sickle cells cannot change shape easily, they tend to burst apart. Normal red blood cells live about 90 to 120 days, but sickle cells last only 10 to 20 days. The body is always making new red blood cells to replace the old cells. However, in sickle cell disease, the body may have trouble keeping up with how fast the cells are being destroyed. Because of this, the number of red blood cells is usually lower than normal. This condition, called anemia, can cause a person to have less energy.
In the United States, most people who have sickle cell disease are of African ancestry or identify themselves as black.
- About 1 in 13 black or African American babies is born with sickle cell trait.
- About 1 in every 365 black or African American babies is born with sickle cell disease.
There are also many people who have sickle cell disease who come from Hispanic, southern European, Middle Eastern, or Asian Indian backgrounds.
About 100,000 Americans have sickle cell disease.
People who do not know whether they carry an abnormal hemoglobin gene can ask their doctor to have their blood tested.
Couples who are planning to have children and know that they are at risk of having a child with sickle cell disease may want to meet with a genetic counselor. A genetic counselor can answer questions about the risk and explain the choices that are available.
Our Team
Meet the team members of the SCD Program

Wally Smith, MD
Director, VCU Adult Sickle Cell Program
Wally Smith, MD
Director, VCU Adult Sickle Cell Program
Internal Medicine
Division of General Internal Medicine
Email: wally.smith@vcuhealth.org

Thokozeni Lipato, MD
Infusion Center Medical Director
Thokozeni Lipato, MD
Infusion Center Medical Director
Internal Medicine
Division of General Internal Medicine

Shirley Johnson, LSW
Program Manager
Shirley Johnson, LSW
Program Manager
Internal Medicine
Shirley is a project manager in charge of supervising the interdisciplinary SCD Adult Medical Home including behavioral health team. She works to reduce hospital re-admissions and length of stay, improve quality and outcomes of care, and improve satisfaction for identified patients.
Phone: (804) 628-2627

Mica Ferlis, ACNP
Nurse Practitioner
Mica Ferlis, ACNP
Nurse Practitioner
Internal Medicine
Mica is a nurse practitioner that works in collaboration with the physicians to deliver health care services to patients with SCD in the inpatient setting.
Email: mica.ferlis@vcuhealth.org

Caitlin McManus, MSN, RN, AGPCNP-B
Nurse Practitioner
Caitlin McManus, MSN, RN, AGPCNP-B
Nurse Practitioner
Internal Medicine
Caitlin is a nurse practitioner that works in collaboration with the physicians to deliver health care services to patients with SCD in the outpatient setting.

Emily Sushko, MSN, RN, AGPCNP-C
Nurse Practitioner
Emily Sushko, MSN, RN, AGPCNP-C
Nurse Practitioner
Internal Medicine
Emily is the nurse practitioner for our outpatient Infusion Clinic, providing urgent and accessible care for our patients. She works in collaboration with the sickle cell team to manage and implement treatment plans for patients who are experiencing an acute sickle cell crisis.
Email: emily.sushko@vcuhealth.org

Kate Osborne, BS, RN
Registered Nurse
Kate Osborne, BS, RN
Registered Nurse
Internal Medicine
Kate is a registered nurse working with our Adult Sickle Cell Disease Team. She has 15 years ICU bedside experience and still works inpatient in the Medical Respiratory ICU a few times a month. Kate is also part of the SCD infusion center team.
Email: kate.sanderson@vcuhealth.org
Justin West, BSN, RN, CMSRN
Registered Nurse
Justin West, BSN, RN, CMSRN
Registered Nurse
Internal Medicine
Justin is a registered nurse who shares outpatient Infusion Clinic and triage nurse duties, providing direct care and helping to address patient questions and concerns. He has also been involved in ongoing efforts to improve care of patients with SCD in the inpatient setting.

Rachel Walls, LCSW
Social Worker

Stefani Vaughan-Sams
Patient Navigator
Stefani Vaughan-Sams
Patient Navigator
Internal Medicine
Stefani is a patient navigators, also working in the clinic every week to assist patients and their families regarding health-related expenses not covered by insurance, transportation costs, and employment options. Patient navigators are also essential in facilitating transition of care.

Nakeyia Williams, BSW
Patient Navigator
Nakeyia Williams, BSW
Patient Navigator
Internal Medicine
Nakeyia is a patient navigators, also working in the clinic every week to assist patients and their families regarding health-related expenses not covered by insurance, transportation costs, and employment options. Patient navigators are also essential in facilitating transition of care.

Marla Brannon, BSW
Patient Navigator
Marla Brannon, BSW
Patient Navigator
Internal Medicine
Marla is a patient navigators, also working in the clinic every week to assist patients and their families regarding health-related expenses not covered by insurance, transportation costs, and employment options. Patient navigators are also essential in facilitating transition of care.
Email: marla.strother@vcuhealth.org

Austin Hardy, CPhT
Prior Authorization Specialist
Austin Hardy, CPhT
Prior Authorization Specialist
Internal Medicine
Austin is a prior authorization specialist. He was hired as a pharmacy tech to handle approvals for opioid medication, check medication fills, incompliance, coordinate with nursing staff on ACC4, and input data for approvals in any medication compliance.

Daniel Sop
Senior Clinical Research Analyst
Daniel Sop
Senior Clinical Research Analyst
Internal Medicine
Daniel is a biomedical engineer who serves as the senior clinical research analyst for the adult SCD program. He uses his engineering training to improve analytical and systematic processes for the SCD program.
Email: daniel.sop@vcuhealth.org

Benjamin Jaworowski
Data & Business Analyst
Benjamin Jaworowski
Data & Business Analyst
Internal Medicine
Ben is a health informatician who is the data and business analyst for the SCD Medical Home. He performs analyses, reporting, and quality improvement for the entire clinical staff.

Donna Casey
Administrative Assistant
Donna Casey
Administrative Assistant
Internal Medicine
Donna facilitates day to day administrative, personnel and program oversight for the Sickle Cell Program Manager and Medical Director. She supports fiscal and time management objectives by coordinating administrative services through effective interactions with the healthcare system team, administration, and support staff as well as hiring, leave and personnel management tasks.
Email: donna.casey@vcuhealth.org
Research
Learn more about our cutting edge research.
Over the course of three decades, the Sickle Cell Disease (SCD) Program at Virginia Commonwealth University (VCU) under the direction of Wally R Smith, MD, Florence Neal Cooper Smith Professor of SCD, has helped lead the national charge for a cure for SCD. By all measures, VCU has become a top 10 Comprehensive Sickle Cell Center in America, caring for nearly 900 children and adults statewide. Further, VCU has helped usher in the dramatic change of SCD from a disease causing death in childhood to one where two-thirds of patients are now adults. This work has included:
- 28 Clinical Drug Trials
- 59 Research Projects
- 82 Symposium Presentations
- 122 Peer Reviewed published papers
- 126 Published abstracts or Proceedings in international venues
- 4 Books, Book Chapters and Monographs
These successes have drawn funding and renown to VCU from government, international scholars, community groups, and leading industry firms in SCD such as The National Institutes of Health, The Health Resources and Services Administration, the Patient Centered Outcomes Research Institute, the New York Academy of Sciences, the Foundation for Sickle Cell Disease Research, Kings College London, Pfizer Pharmaceuticals, Novartis Pharmaceuticals, Global Blood Therapeutics, Emmaus Life Sciences, Johns Hopkins University, and the Food and Drug Administration.
Highlights of the SCD Program include:
- Cure of more than 10 children and adults with SCD using bone marrow transplantation
- Food and Drug Administration approval of Endari (L-glutamine), the second ever anti-sickling drug
- Laboratory development and testing of new potential compounds for the treatment of SCD
- Groundbreaking research on gene therapy to cure SCD
- Groundbreaking, highly cited research on pain in SCD that led to new national definitions surrounding sickle cell pain
- Development of a new evaluation tool and program to help pediatric SCD patients transition to the adult
- A new Medical Home for adults with SCD, consisting of advanced medical, social, psychological, case management, and educational support
- Quicker, smoother Emergency Department and inpatient pain management, customized to the patient
- State Attorney General approval of a regulation that allows physicians to prescribe higher levels of opioids for patients SCD without being forced to justify the treatment to legal watchdogs
The Florence Neal Cooper Smith professorship has supported this groundbreaking work for nearly 5 years, despite being funded at only one quarter of the $1 million dollar endowed chair goal. Our urgent push is to accelerate VCU’s work that will lead to better treatment and a cure for SCD. Over 43 compounds to treat SCD are now in testing, and gene therapy may soon join bone marrow transplant as a cure for SCD. Your dollars can help VCU bring some of these compounds to market, relieve suffering, and bring new hope to patients and families affected by SCD.
To learn more about our current research projects, visit the VCU Sickle Cell Disease Research Program (coming soon!)
Resources
Sickle Cell Disease
- What You Should Know About Sickle Cell Disease
- Using the emergency department with Sickle Cell Disease
- SCD self-care toolkit
- Know your status
- Get Screened
- What you should know about Sickle Cell trait
- Tips for students with Sickle Cell Disease
- SCD and pregnancy
- Living well with SCD in college
- Cure Sickle Cell Initiative
- Sickle Cell Disease for health professionals
- Sickle Cell Disease research, programs and progress
- About Sickle Cell Disease
- Action Medical Research for Children (UK)
- American Sickle Cell Anemia Association: Changing Faces, Changing Shapes (video)
- DNA Learning Center from Cold Spring Harbor Laboratory: Sickle Cell Anemia, 3D Animation with Narration
- Genetic Science Learning Center, University of Utah
- Illinois Department of Public Health
- Information Center for Sickle Cell and Thalassemic Disorders
- MalaCards: sickle cell disease
- Merck Manual Consumer Version
- Michigan Department of Community Health (PDF)
- Nemours Foundation
- Orphanet: Sickle cell anemia
- St. Jude Children's Research Hospital
- University of Rochester Medical Center
- Virginia Department of Health (PDF)
- Washington State Department of Health: Hemoglobin S Fact Sheet (PDF)
- Your Genes Your Health from Cold Spring Harbor Laboratory
- Your Genome from Wellcome Genome Campus
News
Learn more about what's happening in the SCD Program
News Features
- Turning the tide — Sickle cell disease finally gets its due
- Reaching for the sky, Adult Sickle Cell Medical Home transforms sickle cell care
- Social work students find their passion at VCU Health sickle cell program
- Clinical care for sickle cell enters ‘golden age’ after decades on the fringe
- Reducing sickle cell pain — Martin Safo, Ph.D., dedicates his career to sickle cell research
Annual Reports
Contacts
SCD Program
Program Director
Wally Smith, MD
Email: wally.smith@vcuhealth.org
Program Manager
Shirley Johnson, LSW
Phone: (804) 628-2627
Email: shirley.johnson@vcuhealth.org
Administrative Assistant
Donna Casey
Phone: (804) 828-8360
Email: donna.casey@vcuhealth.org